

The modern drug development timeline is highly shaped by manufacturing readiness, analytical understanding, and regulatory alignment. While innovation in therapeutic modalities has accelerated target discovery, the path from laboratory research to approved medicine remains long and complex. In practice, advancing a molecule from discovery to commercialization often exceeds a decade, with delays commonly arising not from clinical outcomes alone but from gaps in early product characterization and quality knowledge.
Increasingly, regulatory agencies expect sponsors to demonstrate a deep understanding of product attributes early in the drug development process, particularly before initiating human studies. Early analytical characterization has therefore become a decisive factor in reducing regulatory uncertainty, avoiding repeated development cycles, and enabling smoother progression through clinical phases.
Why the drug development timeline often exceeds 10 years: structural and regulatory drivers
The length of the drug development life cycle reflects the cumulative complexity of scientific validation, manufacturing scale-up, and regulatory oversight across multiple sequential decision gates. Large-scale analyses of pharmaceutical research and development (R&D) programs show that drug development typically spans 10-15 years from initial discovery to regulatory approval, combining discovery research, preclinical testing, clinical trials, and regulatory review. During this process, attrition is extremely high: thousands of screened compounds enter early discovery, only a few hundred advance to preclinical evaluation.
Several structural factors contribute to extended timelines and rising drug development timelines and costs:
- Iterative optimization of formulation and manufacturing processes
- Evolving regulatory expectations as product knowledge increases
- Scale-up challenges that alter impurity and stability profiles
- Alignment between clinical design and product quality attributes
Regulatory review itself rarely represents the sole bottleneck. Instead, delays frequently emerge when analytical data fail to adequately support product consistency or when manufacturing changes require additional bridging studies. Early-stage decisions that overlook analytical risks often reappear later as regulatory delay in drug approval, forcing sponsors to repeat studies or generate additional data packages.
Furthermore, the need for long-term stability data contributes to the extended timeline for developing a new drug. Any late-stage modification, such as switching from a vial to a pre-filled syringe (PFS), can trigger the need for additional bridging studies or clinical bioequivalence trials, adding years to the development schedule.

Drug development stages and failure rates: how analytical gaps increase attrition
The success rate of drug development by phase reveals a stark reality: approximately 90 % of drug candidates that reach clinical testing ultimately fail. Understanding each phase of drug development is essential to identifying where these failures occur. Phase 1 focuses on safety, Phase 2 on efficacy and side effects, Phase 3 on comparative effectiveness in large populations, and Phase 4 on post-market surveillance.
The highest attrition occurs during the transition to later drug development stages. At phase 3, for instance, the failure rate remains significantly high (over 40 %), frequently driven by a lack of deep molecular understanding rather than a fundamental lack of drug activity.
Analytical gaps commonly lead to failures in the following areas:
- Lack of clinical efficacy (40 % – 50 %): Often, a drug shows efficacy in vitro but fails in humans because developers overemphasize potency while overlooking tissue selectivity.
- Unmanageable toxicity (30 %): This frequently occurs when a drug accumulates in healthy vital organs at a higher rate than in the disease-targeted tissue.
- Poor drug-like properties (10 % – 15 %): Issues such as product instability, high viscosity, or uncontrolled post-translational modifications (PTMs) can compromise safety and potency.
Many of these outcomes originate during the first phases of the drug development process, particularly during candidate optimization and characterization rather than clinical execution itself. Implementing Multi-Attribute Methods (MAM) and high-sensitivity screening protocols (for instance, using ultra-high-performance liquid chromatography) in the early stages enables the elimination of candidates with low specificity or poor tissue selectivity, preventing the costly regulatory delays in drug approval that occur when these issues surface during pivotal trials.
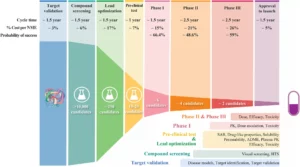
Figure 1. The process of drug discovery and development, and the failure rate at each step. Source: Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022 Jul;12(7):3049-3062.
Preclinical studies in drug development: early characterization before Investigational New Drug (IND) submission
The transition into clinical testing begins with the submission of an IND, which requires sufficient evidence that the investigational product can be consistently manufactured and safely administered. During preclinical studies in drug development, analytical characterization serves as the bridge between discovery science and regulatory evaluation.
Early development activities typically include:
- Definition of a Quality Target Product Profile (QTPP) guiding product attributes
- Establishment of analytical assays to detect degradation and impurities
- Preliminary formulation and stability studies
- Characterization of material used in toxicology studies
As illustrated in development roadmaps, analytical development occurs in parallel with formulation and manufacturing activities rather than sequentially. Establishing fit-for-purpose analytical methods early enables monitoring product changes as manufacturing evolves and ensures that toxicology material remains representative of clinical batches.
Failure to implement early characterization may result in discrepancies between preclinical and clinical materials, prompting regulatory concerns during IND review. Regulatory authorities increasingly encourage early dialogue to clarify expectations, recognizing that better alignment before submission reduces downstream delays.

Accelerating the drug development timeline through early analytical risk mitigation
Speeding up drug development requires a proactive risk mitigation strategy centered on Analytical Quality by Design (AQbD). By establishing a robust in-process control strategy during early development, manufacturers can ensure that the process remains within a proven acceptable range, preventing the drift that leads to batch failures.
The drug development pipeline can be accelerated through several key analytical strategies:
- Early material bridging: Verifying that formulations developed with early-stage material are compatible with the final process material as soon as it becomes available.
- Integrated forced degradation studies: Carrying out photostability and oxidation studies early to understand which residues are susceptible to degradation.
- Digital integrity and traceability: Ensuring all data generated follows strict integrity standards (e.g., 21 CFR Part 11 from the Food and Drug Administration, FDA) to streamline the eventual Biologics License Application (BLA) submission.
Formulation and manufacturing changes (even minor ones such as filter materials or purification adjustments) can significantly influence impurity profiles and stability outcomes. Utilizing advanced analytical platforms like UPLC-MS/MS for impurity profiling and multi-attribute methods (MAM) for critical quality attribute monitoring allows risk anticipation and supports science-based decision-making throughout development.
When these analytical capabilities are implemented early, they help ensure alignment with regulatory expectations and contribute to smoother interactions with health authorities during review.
A well-managed drug development timeline depends on anticipating analytical and quality risks long before they become regulatory obstacles. By integrating advanced characterization strategies early in the drug development process, sponsors can reduce uncertainty, improve IND readiness, and support smoother progression across clinical phases. At AMSbiopharma, we partner with developers to design phase-appropriate analytical strategies, generate reliable characterization data, and help programs move forward with greater regulatory confidence and efficiency.
References
Lai J, Forney L, Brinton DL, Simpson KN. Drivers of Start-Up Delays in Global Randomized Clinical Trials. Ther Innov Regul Sci. 2021 Jan;55(1):212-227. doi: 10.1007/s43441-020-00207-2
Sampathkumar K, Kerwin BA. Roadmap for Drug Product Development and Manufacturing of Biologics. J Pharm Sci. 2024 Feb;113(2):314-331. doi: 10.1016/j.xphs.2023.11.004
Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022 Jul;12(7):3049-3062. doi: 10.1016/j.apsb.2022.02.002
U.S. Food and Drug Administration. The drug development process [Internet]. Silver Spring, MD: FDA; [cited 2026 Feb 23]. Available from: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process