

La planificación en el desarrollo de fármacos está altamente determinada por la fabricación, la comprensión analítica y la alineación regulatoria. Aunque la innovación en modalidades terapéuticas ha acelerado el descubrimiento de dianas, el camino desde la investigación en laboratorio hasta el medicamento aprobado sigue siendo largo y complejo. En la práctica, avanzar una molécula desde el descubrimiento hasta la comercialización a menudo supera una década, con retrasos que comúnmente surgen no solo de los resultados clínicos, sino de lagunas en la caracterización temprana del producto y en el conocimiento de calidad.
Cada vez más, las agencias regulatorias esperan que los patrocinadores demuestren una comprensión profunda de los atributos del producto en las primeras etapas del proceso de desarrollo de fármacos, particularmente antes de iniciar estudios en humanos. Por lo tanto, la caracterización analítica temprana se ha convertido en un factor decisivo para reducir la incertidumbre regulatoria, evitar ciclos repetidos de desarrollo y permitir una progresión más fluida a través de las fases clínicas.
Por qué el cronograma de desarrollo de fármacos a menudo supera los 10 años: impulsores estructurales y regulatorios
El ciclo de vida en el desarrollo de fármacos refleja la complejidad acumulativa de la validación científica, la ampliación de escala de fabricación y la supervisión regulatoria a través de múltiples puntos de decisión secuenciales. Análisis a gran escala de programas de investigación y desarrollo (I+D) farmacéuticos muestran que el desarrollo de fármacos generalmente abarca entre 10 y 15 años desde el descubrimiento inicial hasta la aprobación regulatoria, combinando investigación de descubrimiento, pruebas preclínicas, ensayos clínicos y revisión regulatoria. Durante este proceso, la tasa de abandono es extremadamente alta: miles de compuestos evaluados entran en descubrimiento temprano, y solo unos pocos cientos avanzan a evaluación preclínica.
Varios factores estructurales contribuyen a la extensión de los plazos y al aumento de los tiempos y costes del desarrollo de fármacos:
- Optimización interactiva de la formulación y de los procesos de fabricación
- Expectativas regulatorias en evolución, a medida que aumenta el conocimiento del product
- Desafíos de escala que alteran los perfiles de impurezas y estabilidad
- Alineación entre el diseño clínico y los atributos de calidad del producto
La revisión regulatoria en sí misma rara vez representa el único cuello de botella. En cambio, los retrasos surgen con frecuencia cuando los datos analíticos no respaldan adecuadamente la consistencia del producto o cuando los cambios de fabricación requieren estudios adicionales de comparabilidad. Las decisiones en etapas tempranas que pasan por alto riesgos analíticos, suelen reaparecer más adelante como retrasos regulatorios en la aprobación de medicamentos, obligando a los patrocinadores a repetir estudios o generar paquetes de datos adicionales.
Además, la necesidad de datos de estabilidad a largo plazo, contribuye al extenso plazo para desarrollar un nuevo fármaco. Cualquier modificación en etapas tardías, como cambiar de un vial a una jeringa precargada (PFS), puede desencadenar la necesidad de estudios adicionales de comparabilidad o ensayos clínicos de bioequivalencia, añadiendo años al calendario de desarrollo.

Etapas del desarrollo de fármacos y tasas de fracaso: cómo las lagunas analíticas aumentan la tasa de abandono
La tasa de éxito del desarrollo de fármacos por fase revela una realidad contundente: aproximadamente el 90 % de los candidatos a fármaco que alcanzan la fase clínica finalmente fracasan. Comprender cada fase del desarrollo de fármacos es esencial para identificar dónde ocurren estos fracasos. La Fase 1 se centra en la seguridad, la Fase 2 en la eficacia y los efectos secundarios, la Fase 3 en la eficacia comparativa en grandes poblaciones y la Fase 4 en la vigilancia poscomercialización.
La mayor tasa de abandono ocurre durante la transición a etapas posteriores del desarrollo de fármacos. En la Fase 3, por ejemplo, la tasa de fracaso sigue siendo significativamente alta (más del 40 %), impulsada con frecuencia por la falta de una comprensión molecular profunda más que por una ausencia fundamental de actividad del fármaco.
Las lagunas analíticas conducen comúnmente a fracasos en las siguientes áreas:
- Falta de eficacia clínica (40% – 50%): A menudo, un fármaco muestra eficacia in vitro pero fracasa en humanos porque los desarrolladores enfatizan en exceso la potencia mientras descuidan la selectividad tisular.
- Toxicidad no manejable (30%): Esto ocurre con frecuencia cuando un fármaco se acumula en órganos vitales sanos a una tasa mayor que en el tejido diana de la enfermedad.
- Propiedades farmacológicas deficientes (10% – 15%): Problemas como inestabilidad del producto, alta viscosidad o modificaciones postraduccionales (PTMs) no controladas pueden comprometer la seguridad y la potencia.
Muchos de estos resultados se originan durante las primeras fases del proceso de desarrollo de fármacos, particularmente durante la optimización y caracterización del candidato más que en la ejecución clínica en sí. Implementar Métodos Multiatributo (MAM) y protocolos de cribado de alta sensibilidad (por ejemplo, utilizando cromatografía líquida de ultra alto rendimiento) en etapas tempranas, permite eliminar candidatos con baja especificidad o escasa selectividad tisular, previniendo los costosos retrasos regulatorios en la aprobación de medicamentos que ocurren cuando estos problemas emergen durante los ensayos.
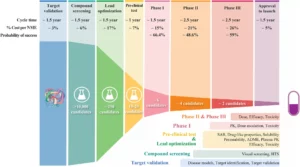
Figura 1. El proceso de descubrimiento y desarrollo de fármacos y la tasa de fracaso en cada etapa. Fuente: Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022 Jul;12(7):3049-3062.
Estudios preclínicos en el desarrollo de fármacos: caracterización temprana antes de la presentación de un nuevo fármaco en investigación (IND)
La transición hacia la evaluación clínica comienza con la presentación de un IND, que requiere evidencia suficiente de que el producto en investigación puede fabricarse de manera consistente y administrarse de forma segura. Durante los estudios preclínicos en el desarrollo de fármacos, la caracterización analítica actúa como puente entre la ciencia de descubrimiento y la evaluación regulatoria.
Las actividades tempranas de desarrollo típicamente incluyen:
- Definición de un perfil de calidad del producto objetivo (QTPP, por sus siglas en inglés) que guíe los atributos del producto
- Establecimiento de ensayos analíticos para detectar degradación e impurezas
- Estudios preliminares de formulación y estabilidad
- Caracterización del material utilizado en estudios toxicológicos
Como se ilustra en las hojas de ruta, el desarrollo analítico ocurre en paralelo con las actividades de formulación y fabricación en lugar de ser secuencial. Establecer métodos analíticos fit-for-purpose desde el inicio permite monitorizar cambios del producto a medida que evoluciona la fabricación y garantiza que el material toxicológico siga siendo representativo de los lotes clínicos.
No implementar una caracterización temprana, puede resultar en discrepancias entre los materiales preclínicos y clínicos, generando preocupaciones regulatorias durante la revisión del IND. Las autoridades regulatorias fomentan cada vez más el diálogo temprano para clarificar expectativas, reconociendo que una mejor alineación antes de la presentación reduce retrasos posteriores.

Acelerando el cronograma de desarrollo de fármacos mediante la mitigación temprana de riesgos analíticos
Acelerar el desarrollo de fármacos requiere una estrategia proactiva de mitigación de riesgos centrada en la calidad analítica por diseño (AQbD, por sus siglas en inglés). Al establecer una estrategia robusta de control en proceso durante el desarrollo temprano, los fabricantes pueden garantizar que el proceso permanezca dentro de un rango aceptable probado, evitando desviaciones que conduzcan a fallos de lote.
El pipeline de desarrollo de fármacos puede acelerarse mediante varias estrategias analíticas clave:
- Puente temprano de materiales: Verificar que las formulaciones desarrolladas con material en etapa temprana sean compatibles con el material del proceso final tan pronto como esté disponible.
- Estudios integrados de degradación forzada: Realizar estudios de fotostabilidad y oxidación desde el inicio para comprender qué residuos son susceptibles a la degradación.
- Integridad y trazabilidad digital: Garantizar que todos los datos generados cumplan con estrictos estándares de integridad (por ejemplo, 21 CFR Parte 11 de la Administración de Alimentos y Medicamentos, FDA) para agilizar la eventual presentación de una Solicitud de Licencia Biológica (BLA).
Cambios en formulación y fabricación (incluso menores, como materiales de filtro o ajustes de purificación) pueden influir significativamente en los perfiles de impurezas y los resultados de estabilidad. El uso de plataformas analíticas avanzadas como UPLC-MS/MS para el perfilado de impurezas y métodos multiatributo (MAM, por sus siglas en inglés) para el monitoreo de atributos críticos de calidad permite anticipar riesgos y respaldar la toma de decisiones basada en la ciencia a lo largo del desarrollo.
Cuando estas capacidades analíticas se implementan tempranamente, ayudan a garantizar la alineación con las expectativas regulatorias y contribuyen a interacciones más fluidas con las autoridades sanitarias durante la revisión.
Un cronograma de desarrollo de fármacos bien gestionado depende de anticipar riesgos analíticos y de calidad mucho antes de que se conviertan en obstáculos regulatorios. Al integrar estrategias avanzadas de caracterización desde etapas tempranas del proceso de desarrollo de fármacos, los patrocinadores pueden reducir la incertidumbre, mejorar la preparación para IND y respaldar una progresión más fluida a lo largo de las fases clínicas. En AMSbiopharma, colaboramos con desarrolladores para diseñar estrategias analíticas apropiadas para cada fase, generar datos de caracterización fiables y ayudar a que los programas avancen con mayor confianza y eficiencia regulatoria.
Referencias
Lai J, Forney L, Brinton DL, Simpson KN. Drivers of Start-Up Delays in Global Randomized Clinical Trials. Ther Innov Regul Sci. 2021 Jan;55(1):212-227. doi: 10.1007/s43441-020-00207-2
Sampathkumar K, Kerwin BA. Roadmap for Drug Product Development and Manufacturing of Biologics. J Pharm Sci. 2024 Feb;113(2):314-331. doi: 10.1016/j.xphs.2023.11.004
Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022 Jul;12(7):3049-3062. doi: 10.1016/j.apsb.2022.02.002
U.S. Food and Drug Administration. The drug development process [Internet]. Silver Spring, MD: FDA; [citado el feb 23 2026]. Disponible en: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process
