

Antes de que un fármaco candidato llegue a la fase de ensayos clínicos, debe demostrar su seguridad y eficacia en condiciones de laboratorio controladas. Este trabajo inicial, conocido como investigación preclínica, determina si un compuesto tiene una probabilidad razonable de éxito en humanos. La calidad y el rigor de estos estudios influyen en la aprobación regulatoria, los plazos de los ensayos clínicos y, en última instancia, la viabilidad comercial del candidato terapéutico.
Para abordar la creciente complejidad de estos estudios, muchas empresas farmacéuticas y biotecnológicas colaboran con organizaciones de investigación preclínica por contrato (CRO) especializadas. Estas colaboraciones proporcionan acceso a experiencia, tecnologías avanzadas y apoyo para el cumplimiento normativo, lo que acelera el cronograma de desarrollo de fármacos. Este artículo explora el alcance de la investigación preclínica por contrato, las mejores prácticas en el diseño de estudios, las principales directrices toxicológicas de la FDA para el cumplimiento y los desafíos comunes que afectan la eficiencia del proyecto.
Definición de la investigación preclínica por contrato: Alcance, ejemplos y situación en la industria
La investigación preclínica abarca todo el trabajo experimental realizado en el desarrollo de fármacos antes de los primeros estudios en humanos, con el fin de comprender cómo se comporta el compuesto en sistemas biológicos y si cumple con los estándares de seguridad necesarios para proceder a los ensayos clínicos.
La investigación preclínica por contrato implica la subcontratación de estas pruebas de fármacos en fase temprana a expertos externos (Organizaciones de Investigación por Contrato, CRO), quienes llevan a cabo diversos estudios, entre ellos:
- Estudios preclínicos en el desarrollo de fármacos, incluyendo la determinación del rango de dosis, toxicología de dosis repetidas, genotoxicidad y farmacología de seguridad.
- Estudios preclínicos en animales para evaluar la tolerabilidad y la toxicidad.
- Desarrollo de métodos para biomarcadores y mediciones farmacocinéticas.
- Documentación regulatoria, incluyendo el informe del estudio preclínico para las solicitudes de autorización de nuevos fármacos en investigación (IND).
Estas organizaciones prestan servicios a empresas biotecnológicas, farmacéuticas y químicas, proporcionándoles datos fiables que fundamentan las decisiones de viabilidad en los procesos de desarrollo.
A medida que crece la demanda mundial de terapias innovadoras, el mercado de empresas de investigación preclínica continúa expandiéndose, con ingresos estimados que se acercan a los 10,5 billones de dólares en 2032, lo que representa una tasa de crecimiento anual compuesto (TCAC) del 7,5 %. Este crecimiento refleja la creciente necesidad de agilidad, integración tecnológica y experiencia regulatoria en las fases preclínicas. Al aprovechar los servicios de las CRO preclínicas, los desarrolladores pueden reducir los costes internos sin comprometer la integridad de los datos ni los plazos de entrega.
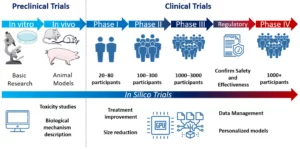
Figura 1. Diagrama de desarrollo de ensayos preclínicos y clínicos, que incluye el tipo de investigación utilizada para la caracterización de cada tratamiento (investigación básica, modelos animales) y el número estimado de participantes en cada fase de los ensayos clínicos. Fuente: Sánchez de la Nava AM et al. Enfermedades cardiovasculares en la era de la salud digital: un enfoque traslacional del laboratorio a la clínica. BioTech (Basel). 30 de junio de 2022;11(3):23.
Diseño de estudios en investigación preclínica: De la planificación a la ejecución
Los programas preclínicos exitosos comienzan con un diseño de estudio proactivo. Las decisiones tomadas en esta etapa influyen en la calidad de los datos, así como en la viabilidad de las presentaciones regulatorias posteriores. De acuerdo con los marcos de desarrollo establecidos, la integración temprana de la farmacología, la toxicología y la farmacocinética en la fase de planificación mejora la capacidad predictiva de los hallazgos no clínicos.
Varios pilares sustentan un diseño de estudio preclínico eficaz:
- Definición clara de objetivos y variables de valoración: los objetivos del estudio deben estar alineados con el plan de desarrollo clínico, la población de pacientes prevista y el mecanismo terapéutico.
- Selección de un modelo biológicamente relevante: la elección de la especie y el modelo debe reflejar las vías metabólicas, la homología de receptores y la fisiología de la enfermedad, cuando corresponda.
- Fundamentación y esquema de dosis: los márgenes de exposición esperados en humanos guían la selección de las dosis iniciales y los patrones de escalamiento.
- Consistencia metodológica: los procedimientos analíticos, el manejo de muestras y los formatos de reporte deben estandarizarse para garantizar la comparabilidad entre estudios.
A lo largo del estudio, la recopilación detallada de datos y el análisis estadístico riguroso fundamentan conclusiones sólidas. La incorporación de elementos de investigación traslacional preclínica, como el modelado farmacocinético/farmacodinámico (PK/PD), mejora las predicciones de las respuestas en humanos. Un diseño de estudio meticuloso reduce los costosos retrasos y respalda las solicitudes regulatorias con evidencia creíble.
Directrices de toxicología de la FDA: Consideraciones para estudios preclínicos
Los organismos reguladores, como la Administración de Alimentos y Medicamentos de los Estados Unidos (FDA), imponen requisitos toxicológicos estrictos que los estudios preclínicos deben cumplir antes de que puedan comenzar los ensayos clínicos. Directrices como la ICH M3(R2), para seguridad general; la ICH S6(R1), para productos biotecnológicos; y la ICH S9, para fármacos oncológicos, especifican cómo deben diseñarse y reportarse los estudios de toxicidad. Los elementos clave de los estudios de toxicología preclínica, según las directrices de la FDA, incluyen:
- Duración del estudio: La toxicología a corto plazo se alinea con los estudios de Fase I, mientras que la toxicidad crónica a largo plazo justifica la realización de ensayos clínicos más extensos.
- Farmacología de seguridad: Las evaluaciones cardiovasculares, respiratorias y neurológicas ayudan a anticipar los efectos adversos en humanos.
- Toxicidad para la reproducción y el desarrollo: Requerida cuando los tratamientos pueden administrarse a pacientes que podrían quedar embarazadas.
- Pruebas de carcinogenicidad: Necesarias para terapias crónicas en las que se prevé una exposición prolongada.

El incumplimiento de estas normas puede conllevar la suspensión o el rechazo de las solicitudes de IND por parte de las autoridades regulatorias, lo que subraya la necesidad de contar con expedientes toxicológicos exhaustivos. La colaboración con socios preclínicos experimentados facilita el cumplimiento normativo y la calidad, a la vez que ayuda a anticipar las expectativas regulatorias.
Cómo superar los retrasos y los desafíos de cumplimiento en las colaboraciones de investigación preclínica
Incluso con requisitos claros, los retrasos en los programas de investigación preclínica por contrato son frecuentes. Suelen producirse cuando la comunicación entre el patrocinador y el socio de investigación es deficiente o cuando los diseños de estudio no están suficientemente alineados con los hitos regulatorios. En este contexto, el papel de un gestor de proyectos cualificado resulta fundamental, ya que garantiza la sincronización entre la ejecución del estudio, los flujos de trabajo analíticos y las estrategias de documentación.

Los desafíos comunes y las formas de mitigarlos son:
- Expectativas desalineadas: Establecer un alcance preciso y criterios de éxito desde el principio.
- Justificación de la dosis incompleta: Vincular los datos farmacológicos y farmacocinéticos con las proyecciones de dosificación desde el inicio.
- Estructura de informes poco clara: Definir plantillas y formatos de entrega durante la planificación.
- Requisitos regulatorios poco considerados: Integrar la revisión de las guías en el diseño del estudio en fase inicial.
Cuando la colaboración funciona eficazmente, las alianzas externas respaldan la toma de decisiones, reducen los riesgos en las vías de desarrollo y permiten a los patrocinadores avanzar con confianza hacia los ensayos clínicos.
El desarrollo preclínico de alta calidad depende tanto del criterio científico y la disciplina en la planificación como de la ejecución experimental. Las organizaciones que priorizan un diseño de estudio coherente, la relevancia traslacional y la alineación regulatoria aumentan sus probabilidades de éxito en la fase clínica.
En AMSbiopharma, apoyamos a las empresas farmacéuticas y biotecnológicas mediante servicios integrados de planificación, ejecución y documentación de estudios preclínicos, combinando el rigor científico con una gestión de proyectos práctica y centrada en los plazos.
Si se está preparando para realizar un trabajo que permita la presentación de una solicitud de autorización para investigación de nuevo fármaco (IND, por sus siglas en inglés) o si está perfeccionando una estrategia de desarrollo en fase inicial, póngase en contacto con nosotros y estaremos encantados de hablar sobre cómo podemos ayudarle.
Referencias
European Medicines Agency (EMA). ICH M3 (R2) Non-clinical safety studies for the conduct of human clinical trials for pharmaceuticals – Scientific guideline [Internet]. Amsterdam: EMA; 2013 Feb 11 [cited 2025 Oct 30]. Disponible en: https://www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals-scientific-guideline
European Medicines Agency (EMA). ICH S9 Non-clinical Evaluation for Anticancer Pharmaceuticals – Scientific guideline [Internet]. Amsterdam: EMA; 2013 Feb 11 [cited 2025 Nov 06]. Disponible en: https://www.ema.europa.eu/en/ich-s9-non-clinical-evaluation-anticancer-pharmaceuticals-scientific-guideline
Huang W, Percie du Sert N, Vollert J, Rice ASC. General Principles of Preclinical Study Design. Handb Exp Pharmacol. 2020;257:55-69. doi: 10.1007/164_2019_277
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Safety guidelines [Internet]. Geneva: ICH; [updated 2024 Aug 30; cited 2025 Nov 06]. Disponible en: https://www.ich.org/page/safety-guidelines
PharmiWeb. Preclinical CRO Market Will Increase USD 10.5 Billion By 2032 [Internet]. 2024 Apr 17 [cited 2025 Oct 30]. Disponible en: https://www.pharmiweb.com/press-release/2024-04-17/preclinical-cro-market-will-increase-usd-105-billion-by-2032
Sánchez de la Nava AM, Gómez-Cid L, Ríos-Muñoz GR, Fernández-Santos ME, Fernández AI, Arenal Á, Sanz-Ruiz R, Grigorian-Shamagian L, Atienza F, Fernández-Avilés F. Cardiovascular Diseases in the Digital Health Era: A Translational Approach from the Lab to the Clinic. BioTech (Basel). 2022 Jun 30;11(3):23. doi: 10.3390/biotech11030023.
U.S. Food and Drug Administration (FDA). S6(R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals [Internet]. Washington (DC): FDA; 2020 March 24 [cited 2025 Oct 30]. Disponible en: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s6-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals