

Before a drug candidate reaches clinical testing, it must demonstrate safety and efficacy under controlled laboratory conditions. This early work, known as preclinical research, establishes whether a compound has a reasonable likelihood of success in humans. The quality and rigor of these studies influence regulatory acceptance, clinical trial timelines, and ultimately, the commercial viability of the therapeutic candidate.
To navigate the increasing complexity of these studies, many pharmaceutical and biotechnology companies collaborate with specialized preclinical contract research organizations (CROs). These partnerships provide access to expertise, advanced technologies, and regulatory compliance support, accelerating the drug development timeline. This article explores the scope of preclinical contract research, best practices in study design, key FDA toxicology guidelines for compliance, and common challenges that impact project efficiency.
Defining Preclinical Contract Research: Scope, Examples, and Industry Situation
Preclinical research is considered all experimental work conducted in drug development before first-in-human studies to understand how the compound behaves in biological systems and whether it meets safety standards to proceed to clinical trials.
Preclinical contract research involves outsourcing these early-phase drug testing to external experts (Contract Research Organizations, CROs), who conduct a range of studies, including:
- Preclinical studies in drug development, including dose-range finding, repeated-dose toxicology, genotoxicity, and safety pharmacology.
- Preclinical studies in animals evaluate tolerability and toxicity.
- Method development for biomarkers and pharmacokinetic measurements.
- Regulatory documentation, including the preclinical study report for investigational new drug (IND) applications
These organizations serve biotech, pharmaceutical, and chemical companies by providing reliable data that drives go/no-go decisions in development pipelines.
As the global demand for innovative therapies grows, the market for preclinical research companies continues to expand, with estimated revenues nearing $10.5 billion by 2032, which represents a Compound Annual Growth Rate (CAGR) of 7.5 %. This growth reflects the heightened need for agility, technological integration, and regulatory expertise in preclinical phases. By leveraging preclinical CRO services, developers can reduce internal costs while maintaining high standards for data integrity and timelines.
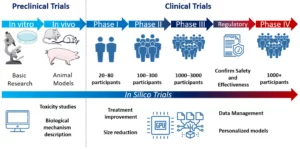
Figure 1. Preclinical and clinical trial development diagram, including the type of research used for the characterization of any treatment (basic research, animal models) and the estimated number of participants for each phase in clinical trials. Source: Sánchez de la Nava AM et al. Cardiovascular Diseases in the Digital Health Era: A Translational Approach from the Lab to the Clinic. BioTech (Basel). 2022 Jun 30;11(3):23.
Study Design in Preclinical Research: From Planning to Execution
Successful preclinical programs begin with proactive study design. Decisions made at this stage influence data quality as well as the feasibility of subsequent regulatory submissions. According to established development frameworks, integrating pharmacology, toxicology, and pharmacokinetics early in the planning phase improves the predictive power of nonclinical findings.
Several pillars support effective preclinical study design:
- Clear definition of objectives and endpoints: study goals must align with the clinical development plan, anticipated patient population, and therapeutic mechanism.
- Biologically relevant model selection: species and model choice should reflect metabolic pathways, receptor homology, and disease physiology when applicable.
- Dose rationale and scheduling: exposure margins expected in humans guide the selection of starting doses and escalation patterns.
- Methodological consistency: analytical procedures, sample handling, and reporting formats need standardization to ensure comparability across studies.
Throughout the study, detailed data collection and rigorous statistical analysis underpin robust conclusions. Incorporating preclinical translational research elements, such as PK/PD modeling, improves predictions of human responses. Meticulous study design reduces costly delays and supports regulatory submissions with credible evidence.
FDA Toxicology Guidelines: Considerations for Preclinical Studies
Regulatory bodies like the US Food and Drug Administration (FDA) impose strict toxicology requirements that preclinical studies must meet before clinical trials can commence. Guidelines such as ICH M3(R2), for general safety, ICH S6(R1), for biotechnology products, and ICH S9, for oncology drugs, specify how toxicity studies should be designed and reported. Core elements of preclinical toxicology studies, according to FDA guidance, include:
- Study duration: Short-term toxicology aligns with Phase I studies, while longer-term chronic toxicity supports extended clinical trials.
- Safety pharmacology: Cardiovascular, respiratory, and neurological evaluations help anticipate adverse effects in humans.
- Reproductive and developmental toxicity: Required when treatments may be given to patients who could become pregnant.
- Carcinogenicity testing: Necessary for chronic therapies where long-term exposure is expected.

Failing to meet these standards can result in regulatory holds or rejection of IND applications, emphasizing the need for thorough toxicology packages. Collaboration with experienced preclinical partners facilitates compliance and quality while helping to anticipate regulatory expectations.
Overcoming Delays and Compliance Challenges in Preclinical Research Partnerships
Even with clear requirements, delays in preclinical contract research programs are common. They often arise when communication between the sponsor and research partner is fragmented or when study designs are not sufficiently connected to regulatory milestones. The role of a skilled project manager becomes central here, ensuring synchronization between study execution, analytical workflows, and documentation strategies.

Common challenges and ways to mitigate them are:
- Misaligned expectations: Establish precise scope and success criteria from the outset.
- Incomplete dose rationale: Tie pharmacology and PK data to dosing projections early.
- Unclear reporting structure: Define templates and deliverable formats during planning.
- Underconsidered regulatory requirements: Integrate guidance review into early-stage study design.
When collaboration functions effectively, external partnerships support decision-making, de-risk development pathways, and enable sponsors to move confidently toward clinical trials.
High-quality pre-clinical development depends as much on scientific judgment and planning discipline as on experimental execution. Organizations that prioritize coherent study design, translational relevance, and regulatory alignment increase their likelihood of successful clinical entry.
At AMSbiopharma, we support pharmaceutical and biotechnology companies through integrated pre-clinical study planning, execution, and documentation services, combining scientific rigor with practical, timeline-focused project management.
If you’re preparing for IND-enabling work or refining an early-stage development strategy, reach out, and we’ll be glad to discuss how we can help.
References
European Medicines Agency (EMA). ICH M3 (R2) Non-clinical safety studies for the conduct of human clinical trials for pharmaceuticals – Scientific guideline [Internet]. Amsterdam: EMA; 2013 Feb 11 [cited 2025 Oct 30]. Available from: https://www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals-scientific-guideline
European Medicines Agency (EMA). ICH S9 Non-clinical Evaluation for Anticancer Pharmaceuticals – Scientific guideline [Internet]. Amsterdam: EMA; 2013 Feb 11 [cited 2025 Nov 06]. Available from: https://www.ema.europa.eu/en/ich-s9-non-clinical-evaluation-anticancer-pharmaceuticals-scientific-guideline
Huang W, Percie du Sert N, Vollert J, Rice ASC. General Principles of Preclinical Study Design. Handb Exp Pharmacol. 2020;257:55-69. doi: 10.1007/164_2019_277
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). Safety guidelines [Internet]. Geneva: ICH; [updated 2024 Aug 30; cited 2025 Nov 06]. Available from: https://www.ich.org/page/safety-guidelines
PharmiWeb. Preclinical CRO Market Will Increase USD 10.5 Billion By 2032 [Internet]. 2024 Apr 17 [cited 2025 Oct 30]. Available from: https://www.pharmiweb.com/press-release/2024-04-17/preclinical-cro-market-will-increase-usd-105-billion-by-2032
Sánchez de la Nava AM, Gómez-Cid L, Ríos-Muñoz GR, Fernández-Santos ME, Fernández AI, Arenal Á, Sanz-Ruiz R, Grigorian-Shamagian L, Atienza F, Fernández-Avilés F. Cardiovascular Diseases in the Digital Health Era: A Translational Approach from the Lab to the Clinic. BioTech (Basel). 2022 Jun 30;11(3):23. doi: 10.3390/biotech11030023.
U.S. Food and Drug Administration (FDA). S6(R1) Preclinical Safety Evaluation of Biotechnology-Derived Pharmaceuticals [Internet]. Washington (DC): FDA; 2020 March 24 [cited 2025 Oct 30]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s6-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals
